Gene module discovery the gold standard missed.
Gene co-expression module discovery is one of the foundational tasks in cancer genomics. WGCNA — the field's most-cited tool, with over 18,000 academic citations — has been the standard since 2008. WGCNA assigns each gene to a co-expression module or to a "background" pool that the algorithm cannot interpret.
Lung adenocarcinoma — GSE31210 (n = 246 patients, 246 dimensions)
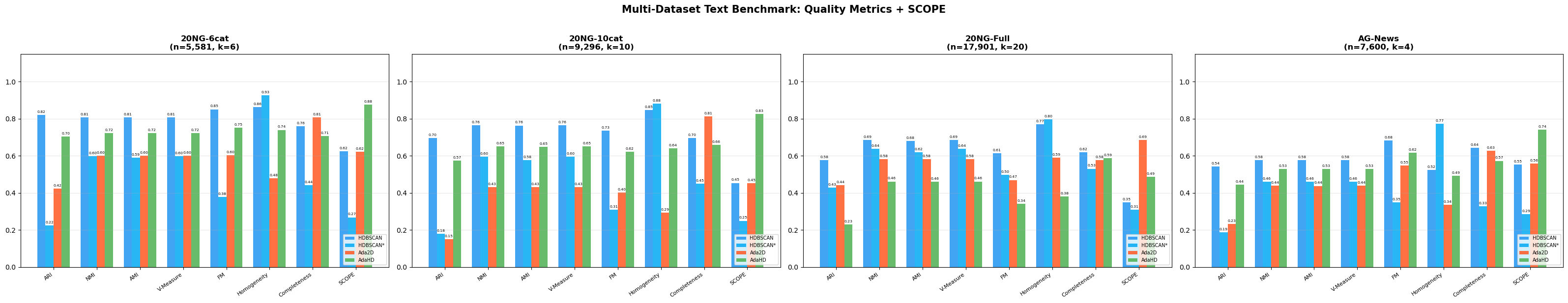
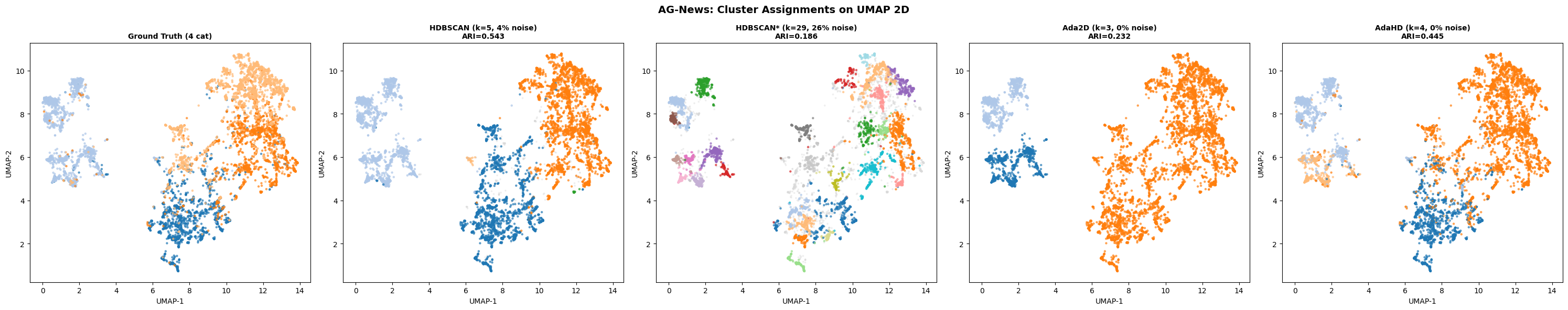
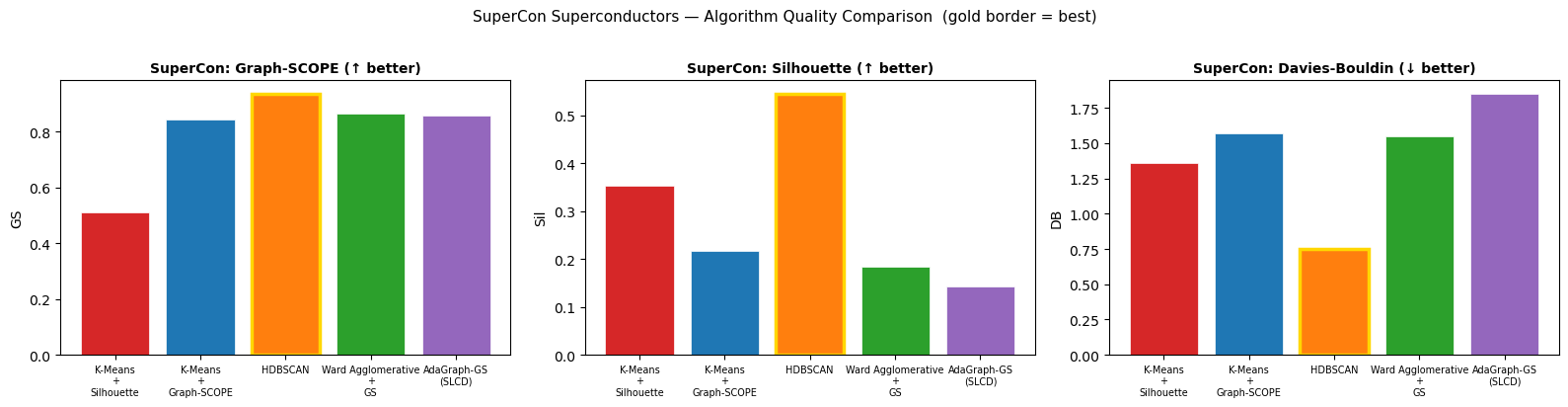
The combined head-to-head scorecard against four established methods (WGCNA, K-Means, Ward, HDBSCAN): 24 wins, 3 ties, 13 losses across 40 head-to-head comparisons spanning multiple quality and biological-coherence metrics. Most importantly: WGCNA placed 76.3% of genes into its uninterpretable "background" pool. AdaGraph clustered 100% of genes into 12 modules.
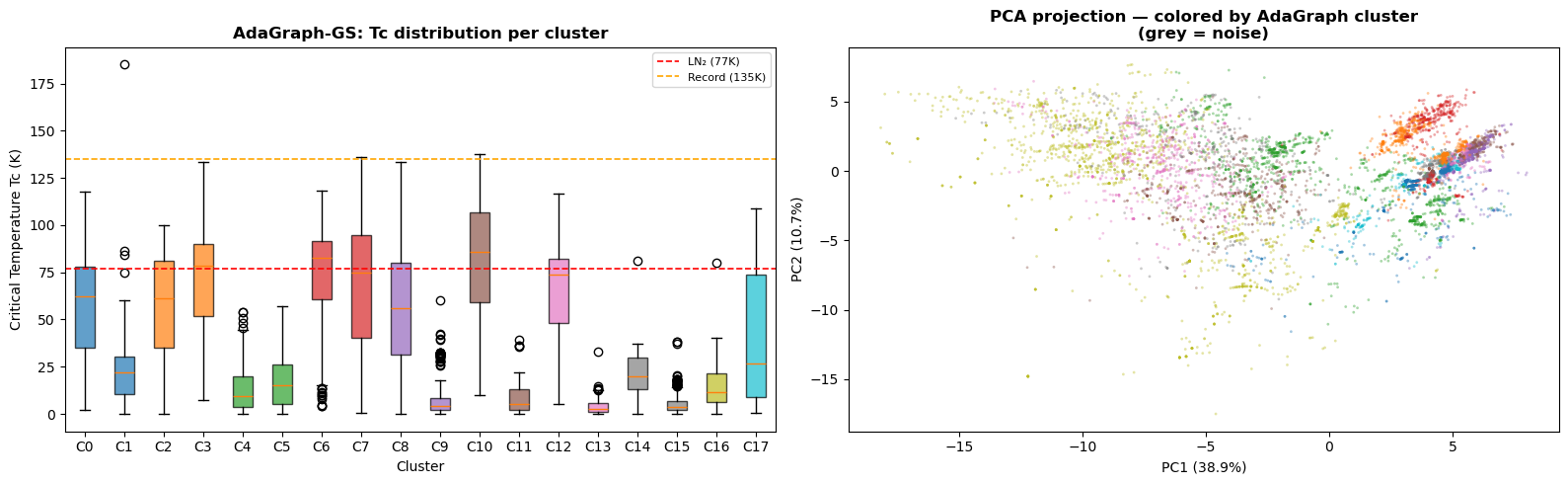
AdaGraph identified a 44-gene smoking-relapse interaction module (cluster C6) in the lung cancer dataset that WGCNA distributed across its background pool, recovering only 2 of the 44 genes. An independent literature validation of every gene-cluster assignment found zero contradictions with established cancer biology, and identified 5 novel candidate genes for experimental follow-up: LOC101060363, TMA7, IRF2BPL, RPS4XP2, MIR6805.
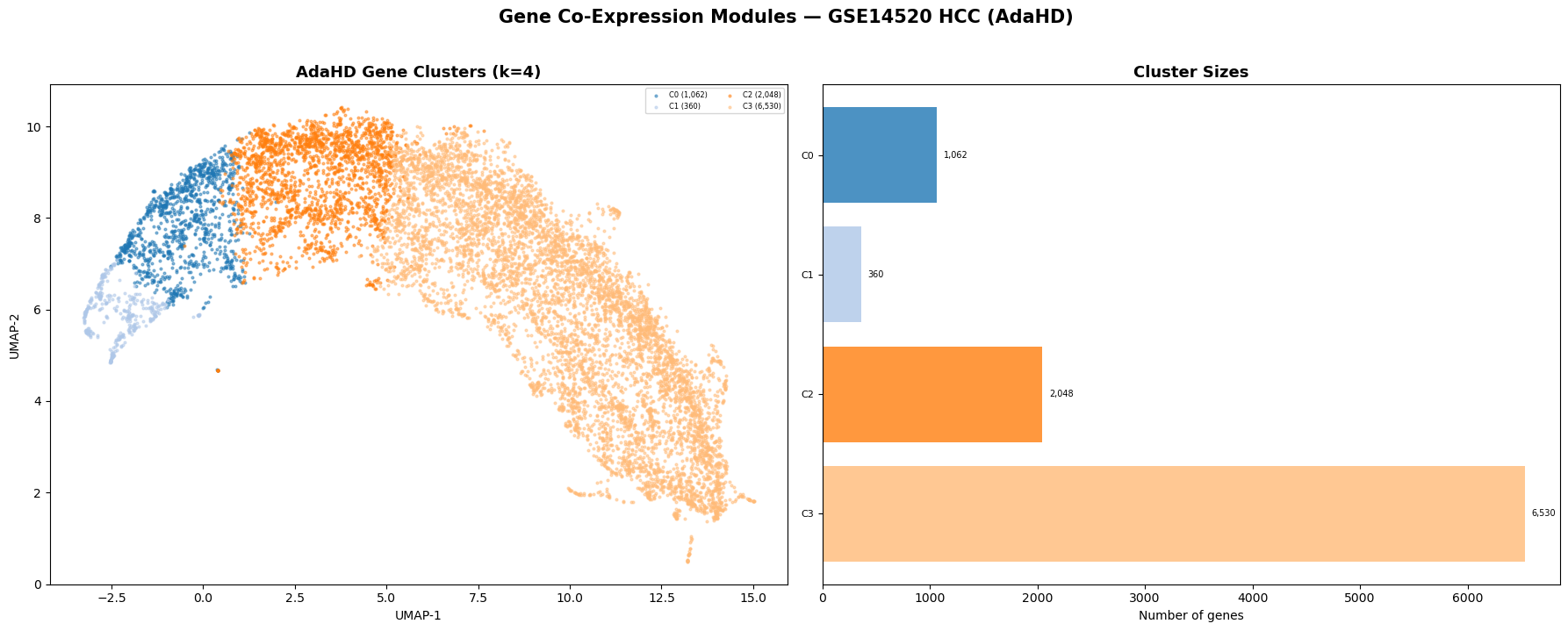
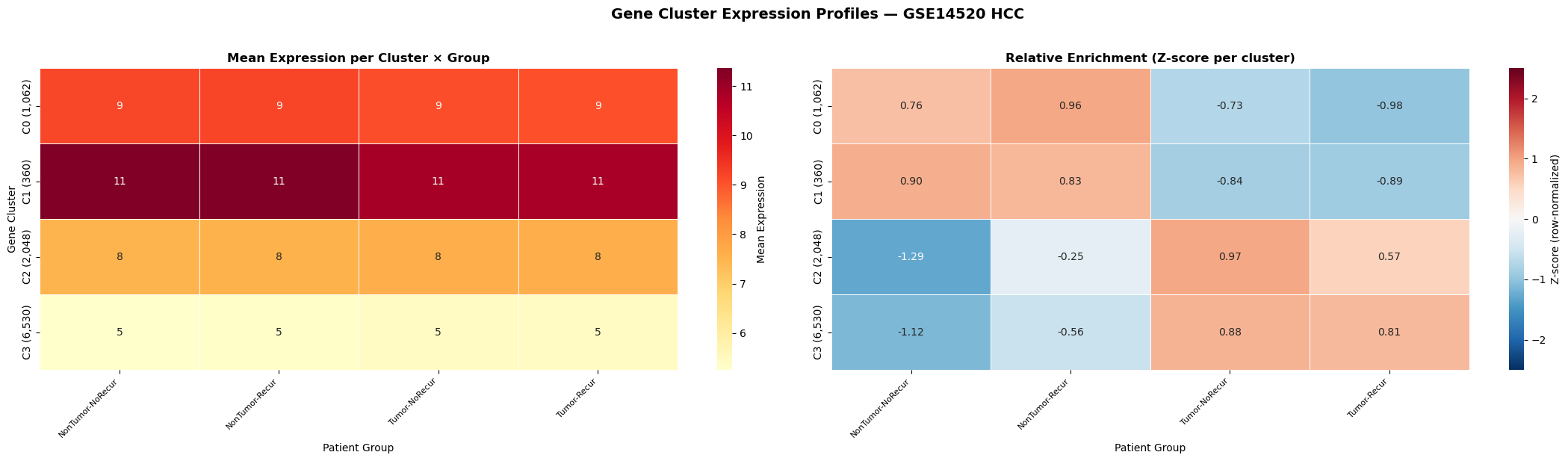
Hepatocellular carcinoma — GSE14520 (n = 431 patients, 10,000 genes)